CAP science
CAP is a consortium with activities in scientific research in chemistry, physics and biology. What we have in common is a vision that we can create green and sustainable fuels and chemicals, using solar energy and other renewables. A few of our scientific research publications from 2023 are listed below. To view titles from previous years, select a year in the menu above, or click 2021 or 2022.
Picks of our best science in 2023!
February 2023:
Rhys Grinter, Ashleigh Kropp, Hari Venugopal, Moritz Senger, Jack Badley, Princess R. Cabotaje, Ruyu Jia, Zehui Duan, Ping Huang, Sven T. Stripp, Christopher K. Barlow, Matthew Belousoff, Hannah S. Shafaat, Gregory M. Cook, Ralf B. Schittenhelm, Kylie A. Vincent, Syma Khalid, Gustav Berggren & Chris Greening published an article in Nature:
Structural basis for bacterial energy extraction from atmospheric hydrogen.
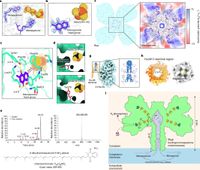
Diverse aerobic bacteria use atmospheric H2 as an energy source for growth and survival. This globally significant process regulates the composition of the atmosphere, enhances soil biodiversity and drives primary production in extreme environments. Atmospheric H2 oxidation is attributed to uncharacterized members of the [NiFe] hydrogenase superfamily. However, it remains unresolved how these enzymes overcome the extraordinary catalytic challenge of oxidizing picomolar levels of H2 amid ambient levels of the catalytic poison O2 and how the derived electrons are transferred to the respiratory chain. Here we determined the cryo-electron microscopy structure of the Mycobacterium smegmatis hydrogenase Huc and investigated its mechanism. [The] findings provide a mechanistic basis for the biogeochemically and ecologically important process of atmospheric H2 oxidation, uncover a mode of energy coupling dependent on long-range quinone transport, and pave the way for the development of catalysts that oxidize H2 in ambient air.
February 2023:
João S. Rodrigues, Barbara Bourgade, Karen R. Galle & Pia Lindberg published an article in Microbial Cell Factories: